
Actividad metabólica del tejido adiposo y su relación con la resistencia a la insulina
Abstract
Numerosas explicaciones fisiopatológicas del síndrome metabólico han propuesto que la resistencia a la insulina, la inflamación crónica y la distribución ectópica del tejido adiposo son manifestaciones metabólicas de la actividad del tejido adiposo en respuesta a los cambios en el suministro y tipo de energía constitutiva de la alimentación diaria. La paradoja propuesta es la «falla en la función del tejido adiposo» que se manifiesta con una clara expresión sistémica de la disfunción como lo son las lipodistrofias que ahora son más comunes en la población. La piedra angular del modelo fisiopatológico de la «falla del tejido adiposo» es la causa más clara hasta el momento de la resistencia a la insulina. Teniendo en cuenta la función endocrina del tejido adiposo que contribuye con la secreción de leptina, adiponectina, resistina, interleukina 6, Factor de necrosis tumoral α, y visfatina, convierten al tejido adiposo en una glándula endocrina y no en una mera bodega de reserva como se definió por largo tiempo.
La información hasta la fecha sobre las secreciones anteriormente mencionadas es incompleta, pero han podido relacionar con cierto grado de evidencia científica la contribución de estos factores con la resistencia a la insulina y el síndrome metabólico. En la presente revisión se describirán las actividades metabólicas del tejido adiposo y su relación con la resistencia a la insulina teniendo en cuenta las bases moleculares, bioquímicas y fisiológicas de la patología más diagnosticada en la última década.
Palabras Claves
Metabolismo, insulina, síndrome metabólico, tejido adiposo.
La importancia del tejido adiposo dentro de la especies presenta una gran variabilidad, es así como en los invertebrados el tejido adiposo representa un órgano importante en los insectos, pero cuantitativamente su importancia decrece en los arácnidos, crustáceos y moluscos. En los vertebrados el desarrollo del tejido adiposo es un signo evolutivo en los organismos homeotermos, también su proporción en el peso corporal varia notoriamente entre las especies (cerca del 40% del peso corporal de los cetáceos lo constituye el tejido adiposo) las reservas grasas de las aves migratorias y las reservas de los mamíferos también presentan gran diferencia, pero dan muestra de la importancia en la actividad metabólica de este órgano. El desarrollo del tejido adiposo blanco que es el tipo de tejido en el que se centrar esta revisión también presenta amplias diferencias entre las especies. Este no puede ser detectado microscópicamente durante la etapa embrionaria ni en el nacimiento de la mayoría de los roedores (ratones y ratas), mientras que si está presente en el nacimiento de los conejos, cerdos, conejillos de indias y en los humanos (1,2).
Histológicamente el tejido adiposo blanco parece estar bien vascularizado, muchos adipocitos están en contacto con un capilar. Estos surten adecuadamente la entrada y salida activa de metabolitos y secreciones de varios péptidos y factores no peptídico. Adicionalmente células endoteliales como los fibroblastos y otras células de origen mesenquimal incluyendo los preadipocitos y los adipocitos como tal, secretan factor de crecimiento similar a la insulina (IGF-1) lo que sugiere que este es el principal protagonista de la hiperplasia del tejido adiposo durante la embriogénesis y en la infancia.
Existen dos picos de crecimiento acelerado del tejido adiposo blanco, uno después del nacimiento y otro entre los 9 y 13 años (3). La tasa de proliferación del tejido adiposo decrece en la adolescencia y se presenta un relativo equilibrio o estabilidad en el peso corporal; las células adipocitarias mantienen en número estable hasta la adultez. Una expansión del tejido adiposo se debe principalmente a una hipertrofia de las células ya presentes hasta el momento y no hay aumento en el número de células. Solamente en casos de obesidad extrema se han reportado aumento en el número de las células en más o menos tres veces (4).
Algunos desordenes endocrinos definidos indican que algunas hormonas pueden afectar la masa y el patrón de distribución del tejido adiposo (tabla 1). Haciendo énfasis en la actividad metabólica del tejido adiposo y los determinantes endocrinos de una posible caracterización de la obesidad, la regulación del balance lipolítico/lipogenético y la fisiología lipogenético y la fisiología hormonal demarcaran el camino del tejido adiposo proliferante.
Tabla 1: Efecto de algunas hormonas, Citokinas y factores de crecimiento sobre la celularidad del tejido adiposo en estudios in vivo e in vitro. (5)
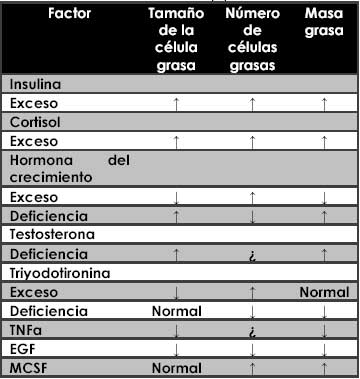
MCSF: Macrophage colony-stimulating factor, TNFα: Tumor Necrosis Factor, EGF: Epidermal Growth
Los adipocitos son capaces de expresar y secretar un número considerable de hormonas, Citokinas y péptidos que ayudan al mantenimiento de la homeostasis; péptidos vasoactivos cuyos productos proteolíticos regulan el tono vascular; la leptina que tiene un rol central en la regulación del balance energético. También son capaces de producir hormonas esteroideas activas, incluyendo estrógenos y cortisol, por la conversión de los precursores androgénicos y los glucocorticoides inactivos respectivamente (6).
A través de los anteriores productos, los adipocitos pueden influenciar de manera muy importante la biología adipocitaria local en el organismo humano, así como también el metabolismo sistémico de diversos sitios como el cerebro, hígado, músculos, las células β del páncreas, las gónadas, los órganos linfoides y la vasculatura sistémica (7).
El tejido adiposo está estrechamente regulado por la actividad metabólica de muchas hormonas. Cada hormona tiene un efecto peculiar de acuerdo a la expresión de su respectivo receptor y esto varía según la edad y el género y expresa un comportamiento diferente en la distribución del tejido adiposo. Estas particularidades regulan dos procesos notoriamente importantes como lo son la acumulación del tejido adiposo (lipogénesis) y su utilización y gasto (lipolisis). Hay también muchas diferencias entre estos dos procesos en posiciones anatómicas diferentes como lo es la grasa subcutánea y la grasa visceral. Estos efectos depende en gran medida de la actividad de la lipoprotein lipasa (LPL) y lipasa hormona sensible (HSL).
Funciones del Tejido Adiposo: Lipogénesis y Lipólisis
LPL: Lipoprotein lipasa: Lipogenesis
La LPL es una enzima extrahepatica responsable de la hidrólisis de los triacilgliceroles de los quilomicrones y de las lipoproteínas de muy baja densidad (VLDL) a manera de control y paso limitante en el aclaramiento de los ácidos grasos libres de la sangre. Esta enzima está localizada principalmente en la superficie endotelial a nivel del tejido adiposo y tejido muscular igualmente. La insulina, los glucocorticoides, las catecolaminas, la interleukina 6 y 1, junto con otros factores diferentes, modulan la actividad de la enzima en los depósitos de grasa. Esta enzima muestra una diferencia marcada entre los géneros, esto debido a la diferencia en la expresión de los receptores en los adipocitos. En los hombres la actividad de la LPL, medida por triglicéridos radiomarcados, es 50% mayor en el omentum que en la grasa subcutánea abdominal. Contrariamente en las mujeres muestra un comportamiento distinto, en ellas los adipocitos del omentum son más pequeños y la actividad de la LPL es mucho menor en el tejido adiposo abdominal que en la grasa subcutánea, contribuyendo de esta manera a una disposición mayor a padecer de obesidad [8].
Lipasa Hormona Sensible (HSL): Lipólisis
El tejido adiposo libera energía en respuesta a las demandas del cuerpo. La lipólisis en el tejido adiposo ha sido ampliamente estudiada in vivo e in vitro porque son fácilmente cuantificable los productos finales como los ácidos grasos libres y el glicerol inducidos por este proceso de lipolisis. Esta hormona específica unida a los receptores de membrana plasmáticos inicia la cascada lipolitica. Estos receptores se ligan a las proteínas G que presentan dualidad en su función, donde pueden ser estimuladoras Gs o inhibidoras Gi. Las proteínas G son capaces de activar intracelularmente un medio catalítico mediado por Adenilciclasa convirtiendo el ATP en AMP cíclico. Este AMP cíclico se une a la subunidad reguladora de la proteinkinasa, liberando así las subunidades catalíticas que fosforilaran y activaran la HSL. Solo la insulina y las catecolaminas poseen efectos agudos sobre la lipolisis. La insulina inhibe la lipolisis [9]. Mientras las catecolaminas pueden estimular los receptores β y pueden inhibir los receptores α2 [10]. La actividad de la HSL se puede incrementar por la ACTH, el glucagon, la TSH, el AMP cíclico, la cafeína y la teofilina.
Fisiología de la Regulación Hormonal en la Distribuición del Tejido Adiposo
Los adipocitos son células que responden a los estímulos de la insulina. Esta hormona es un regulador virtual de todos los procesos y secuencias metabólicas de la fisiología del adipocito. La insulina promueve el almacenamiento de los triacilgliceroles en los adipocitos por varios mecanismos, donde se incluyen el fomento en la diferenciación de preadipocitos a adipocitos y estimulando la síntesis y el transporte de la glucosa y los triacilgliceroles, así como también inhibiendo la lipolisis en los adipocitos maduros o adultos.
Los efectos metabólicos de la insulina son mediados por cambios extremadamente veloces en la fosforilación y función de las proteínas, también como en cambios en los target de la expresión génica. La insulina y los glucocorticoides representan el principal estimulante fisiológico de la actividad de la LPL; esta asociación juega un rol importante en la topografía de la grasa corporal. Cuando el tejido adiposo del omentum, resistente a la actividad de la insulina, es expuesto a una combinación de insulina y dexamenthasona por 7 días, se presenta un incremento en el mRNA de la LPL [11]. GLUT 4 es el principal transportador de glucosa a nivel celular, localizado primariamente en las células musculares y en los adipocitos (12,13). En presencia de la insulina, GLUT 4 es translocado del espacio intracelular a la membrana plasmática permitiendo así el transporte de glucosa hacia el interior de la célula (13). La señal molecular inicial para la acción de la insulina involucra la activación del receptor de substrato de insulina (IRSs) por sus siglas en inglés, la activación del fosfoionositide 3´kinasa (PI3K) que permiten las translocación de GLUT 4 a la membrana plasmática (14, 15,16).
La insulina también juega un papel importante en la secreción de la leptina, aunque la literatura actual aun no concuerda en este hecho. Se ha demostrado que a diversas formas de administración de insulina a nivel agudo (horas) como a nivel crónico (días) in vivo e in vitro se incrementa el mRNA o (gen de la obesidad) en roedores y en humanos (17, 18, 19,20).
Contradictoriamente Sinha y colaboradores no encontraron correlación alguna entre los perfiles de 24 horas entre insulina y leptina en términos de concentración (21). Lo cual no quiere decir que la hiperinsulinemia, característica principal en la obesidad, pueda mantener unos niveles altos de expresión de la leptina. El comportamiento es variable en condiciones medioambientales y por asociaciones endocrinas que predisponen al organismo a responder frente a la descarga hormonal que termina por acentuar la expresión del comportamiento fisiopatológico de la obesidad y del síndrome metabólico.
La insulina también desempeña un rol favorable en la activación de la LPL mediado por el cortisol. Estas combinadas, describen el comportamiento de la LPL en términos de su actividad en los depósitos de grasa a nivel visceral, subcutánea, considerando que la grasa abdominal muestra una mayor densidad en los receptores para los glucocorticoides, lo que puede diagramar una hipótesis en la lipodistrofia que se presenta en patologías como el SIDA (tratamiento con antirretrovirales) y en la enfermedad de Cushing en donde la terapia con glucocorticoides manifiesta este comportamiento (22).
Para no sesgar esta revisión sistemática y la opinión crítica del lector se tendrán en cuenta todos los factores hasta la fecha descritos en la fisiología hormonal de la distribución del tejido adiposo en el organismo humano. En aras de enfocar la revisión a la resistencia a la insulina y obviamente al síndrome metabólico, se describió anteriormente el rol de la insulina en toda la fisiología y la consecuente patología. Las demás hormonas y los otros factores puede ver su acción resumida en la tabla # 2.
Tabla 2: hormonas y otros factores asociados a la distribución del tejido adiposo y sus correspondientes mecanismos de acción (23).
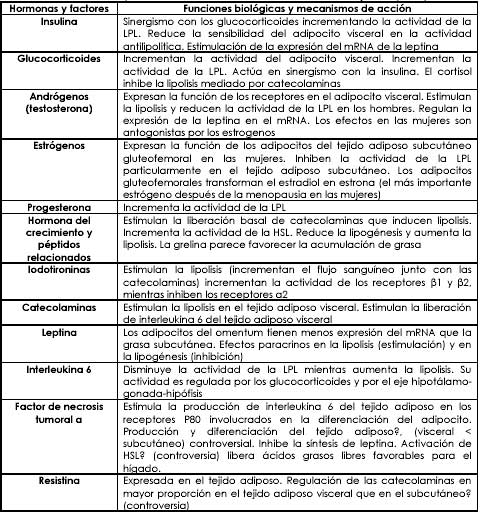
Adaptado de: Endocrine Determinants of Fat Distribution. Handbook of obesity. Etiology and pathophysiology. 2006. New York
Resistencia a la Insulina
La resistencia a la insulina es un estado patológico común en donde las células objetivo de esta hormona fallan en responder a las concentraciones plasmáticas de esta. Esto resulta en la inhabilidad de la hormona para mantener la homeostasis glicémica y lipídica en el organismo. Por lo tanto hay una demanda en el aumento de la producción de la insulina para compensar la hiperglicemia y la falta de sustrato glicémico en las células glucosadependientes (24,25). Las principales características de la resistencia a la insulina es la desinhibición de la lipolisis en el tejido adiposo, la falta de utilización de la glucosa en las células dependientes de glucosa y la desinhibición de la gluconeogenesis. Los marcadores clínicos de la resistencia a la insulina son: la obesidad visceral, acantosis nigricans (26), acné, hirsutismo (27), y esteatosis hepática (24).
La resistencia la insulina está asociada con un gran número de patologías entre las cuales se destacan: la obesidad, el síndrome metabólico, la diabetes mellitus tipo 2, lipodistrofias, síndrome de ovario poliquístico e infecciones crónicas. La prevalencia general de la resistencia a la insulina hasta ahora reportada es de 10- 25% (28).
La etiología de la resistencia a la insulina incluye factores genéticos que resultan en diversas formas sindromicas de resistencia a la insulina, junto con factores ambientales como: la alimentación, la inactividad física, la edad, el consumo de cigarrillo y el uso de medicamentos entre los cuales se encuentran las tiazidas (diuréticos), antagonistas beta-adrenérgicos y glucocorticoides (26). El factor más importante es la obesidad sin duda alguna, que usualmente esta combinado con factores poligenéticos y ambientales (26,29).
La resistencia a la insulina hace que el tejido adiposo visceral se vuelva resistente a la acción antilipolitica de la insulina y como consecuencia se comienzan a liberar cantidades excesivas de ácidos grasos libres. Esta reacción es la que inicia todo el perfil patológico sistémico de la resistencia a la insulina. También, hay liberación de varias hormonas y citokinas del tejido adiposo (adipokinas) que contribuyen a los efectos metabólicos y el comportamiento bioquímico del tejido adiposo descrito en detalle en la tabla # 2.
Mecanismos de la Resistencia a la Insulina
1. Desbalance en la secreción de ácidos grasos libres y adipokinas
Se ha establecido claramente que la secreción inadecuada de ácidos grasos libres provenientes del tejido adiposo juega el rol más importante en la patogenia de la resistencia a la insulina (30). Numerosos estudios han encontrado asociación directa entre la resistencia a la insulina y el incremento en los niveles del factor de necrosis tumoral α, (TNF-α), interleukina 6 (IL-6), Proteína quemoatrayente 1 de macrófagos y monocitos (MCP-1), Inhibidor-1 de la activación del plasminogeno (PAI-1), adipsina y una disminución marcada de adiponectina (31).
2. Ácidos grasos libres y TNF-á
El factor de necrosis tumoral α, es el principal factor autocrino/paracrino que favorece la secreción de ácidos grasos libres del tejido adiposo a la circulación general (32). Sin embargo, aún se desconoce la causa o el factor que favorece la secreción de adipokinas del tejido adiposo. El TNF-α media la represión de varios genes responsables de la utilización y almacenamiento de la glucosa y los ácidos grasos. La actividad del TNF-α aumenta la rata de lipolisis en el tejido adiposo favoreciendo así la liberación de ácidos grasos libres y citokinas, comportamiento etiológico de la resistencia a la insulina. El TNF-α y los ácidos grasos libres, perjudican la señalización de la insulina en los tejidos insulino dependientes, principalmente el tejido muscular. Esta liberación de ácidos grasos libres, de acuerdo con la hipótesis de Randle, actúan predominantemente en el metabolismo intermedio. Incrementando el NADH/NAD+ y el radio de acetyl CoA/CoA, lo cual es una de las posibilidades de la preferencia en la utilización de los ácidos grasos libres como substrato por encima de la glucosa [33,34]. El tejido adiposo visceral secreta estos ácidos grasos libres directamente al torrente sanguíneo donde sus efectos más inmediatos los empieza a sufrir el hígado en contraste con el tejido adiposo subcutáneo, haciendo que la actividad metabólica del hígado este sobrecargada de una manera que puede llegar a desencadenar en muy poco tiempo patologías como la esteatosis hepática e hígado graso, debido al suministro constante de ácidos grasos endógenos muchas veces acompañados por ácidos grasos exógenos (provenientes de la dieta).
En el hígado, los altos niveles de ácidos grasos libres promueven bastante substrato para la síntesis de los triacilgliceroles, las lipoproteínas de muy baja densidad VLDL y para la gluconeogenesis. El TNF-α reprime los genes de la utilización de la glucosa y de la β-oxidación (35). La síntesis de novo de colesterol ocurre gracias a la estimulación que ejerce el TNF-α en la activación de los genes correspondientes. Los ácidos grasos libres también aumentan la síntesis de fibrinógeno y PAI-1 en el hígado (36).
En el musculo las altas concentraciones de ácidos grasos libres favorecen la β- oxidación con lo que se disminuye la tasa de captación y de utilización de la glucosa (37). Pero aun así la β-oxidación es incapaz de realizar un óptimo aclaramiento plasmático de los ácidos grasos libres y mucho más aún si hay una inactividad física (38). La síntesis de glucógeno también se ve inhibida por estos factores. Teniendo en cuenta que el tejido muscular es el principal consumidor de glucosa en el cuerpo (80- 90%) la disminución en la utilización de la misma contribuye a la hiperglicemia (39). Este exceso de ácidos grasos libres es acumulado como gotas de triacilgliceroles en el musculo.
En el tejido adiposo, los ácidos grasos libres inhiben la actividad de la LPL, como ya lo había mencionado anteriormente, que por consiguiente estimulara a la insulina lo que hará aún más imposible el aclaramiento de los ácidos grasos libres en el plasma (40).
En las células β la exposición prolongada a los ácidos grasos libres desequilibra la secreción de insulina, lo cual es la manifestación más clara de la diabetes mellitus tipo 2.
El TNF-α ha demostrado poseer actividad en la regulación de la actividad de la adiponectina, en los receptores GLUT 4, en el receptor de substratos de insulina 1 (IRS-1), en la proteína estimulante del ligando CCA AT α, en el receptor activante y proliferador del peroxisoma gamma, y la perilipina en los adipocitos. El TNF-α regula la expresión de algunos genes en el tejido adiposo que son responsables de la inflamación, la respuesta inmune y el gasto energético (molécula de adhesión celular vascular 1 VCAM 1, plasminogeno activador/inhibidor 1, IL 6, IL 1-β, angiotensinogeno, resistina y leptina (34).
3. Adiponectina
La adiponectina está inversamente relacionada con la resistencia a la insulina en la lipodistrofia, obesidad y en estados de inflamación (41,42). La adiponectina promueva la sensibilidad a la insulina por varios mecanismos. En el hígado, esta induce la oxidación de ácidos grasos libres, reduce su síntesis y disminuye la utilización de estos y reprime la gluconeogenesis por una regulación enzimática (43,44). Los niveles de adiponectina se incrementan cuando se presenta una pérdida de peso (45) y con el tratamiento con thiazolidinedionas (46).
La síntesis de adiponectina se desequilibra en estados de exceso calórico, lo cual es asociado también con resistencia a la leptina o a una deficiencia. El factor de crecimiento similar a la inulina 1 (IGF-1) estimula la síntesis de adiponectina (43). La adiponectina ejerce sus efectos sobre los factores de transcripción génica y también a través de la inhibición del factor de transcripción nuclear Kappa-β y este a su vez se activa a través del TNF- α (46). La adiponectina también disminuye la expresión de las moléculas de adhesión en la pared de los vasos sanguíneos, inhibiendo la quemotaxis de los macrófagos y su conversión a células espumosas, la proliferación de las células de musculo liso y los eventos inflamatorios en la aterogénesis también se ven inhibidos en presencia de la adiponectina, estos son promovidos principalmente por la IL-6 y PAI-1. Adicionalmente la adiponectina suprime la secreción de TNF-α (47).
La Obesidad es el Principal Factor de la Resistencia a la Insulina
Todas las personas tienen su peso determinado genéticamente, este peso a su vez está estrechamente regulado por mecanismos energéticos homeostáticos. Continuando con la discusión del inicio de esta revisión se observa que la tendencia a creer que la leptina y la insulina son secretadas en igual proporción dependiendo de la cantidad de tejido adiposo, es apoyada con un mayor número de adeptos que están de acuerdo con la hipótesis. A largo plazo la leptina y la insulina terminan por ser los reguladores de la masa corporal del hombre.
La obesidad así se convierte en el principal protagonista de la resistencia a la insulina puesto que favorece todos los medios por los cuales el tejido adiposo expresa todo su potencial genético respaldado por los factores ambientales, son la piedra angular de la actividad metabólica del tejido adiposo (48, 49,50).
Conclusión
La importancia del tejido adiposo es ahora más notoria en términos del comportamiento homeostático del organismo humano. Ya no es considerado como la despensa o la bodega orgánica de energía altamente utilizable como el substrato graso, y pasa a ser un órgano totalmente endocrino que es capaz de mediar y adaptarse a los cambios fisiológicos de la secreción de un gran número de hormonas y factores bioquímicos relacionados con la obesidad. La resistencia a la insulina es una patología realmente simple que está estrechamente relacionado con la actividad metabólica del tejido adiposo que se revisó en este artículo. El desconocimiento de la etiología de la enfermedad hace que el tratamiento sea más lento, costoso y poco efectivo, teniendo en cuenta los métodos de acercamiento al tratamiento de la obesidad y el sobrepeso que actualmente son más estéticos, relegando el componente bioquímico. Las oportunidades para el tratamiento de la resistencia a la insulina, el síndrome metabólico y la obesidad son innumerables ya que existe la posibilidad de inhibir o reprimir varias vías de señalización celular que modificarían la patogenia de este trío de condiciones fisiológicos que afectan a un número más elevado de personas en la actualidad. La alternativa más fácil para optimizar la sensibilidad de las receptores celulares que permiten la entrada de glucosa mediada por insulina a la célula es tan solo aumentar la actividad física y el gasto energético, disminuir el consumo excesivo de calorías y distribuir correctamente los sustratos alimentarios que pasaran a ser el combustible celular. El tratamiento de estas condiciones patológicas debe realizarse en compañía de un grupo interdisciplinario de profesionales en salud y el paciente debe partir de la base que requiere del acompañamiento de expertos en el área y lo más importante es que debe abstenerse de utilizar medios alternativos como el referimiento de píldoras, batidos o suplementos de cualquier tipo. Después de haber conocido concretamente la intricada red de funciones metabólicas del tejido adiposo y su relación con la resistencia a la insulina el lector debe al menos comprender la magnitud bioquímica de su patología y suponer que el tratamiento debe realizarlo un profesional competente con experiencia en el área. Lo más importante y lo que siempre recalco en las conclusiones de todos mis escritos es que cualquier procedimiento médico debe tener un alto bagaje científico y debe ser basado en la evidencia médica.
Notas
- Hausman GJ. Identification of adipose tissue primordial in perirenal tissues of pig fetuses: utility of phosphatase histochemistry. Acta Anat 1987; 128: 236-242
- Poissonnet CM, Burdi AR, Garn SM. The chronology of adipose tissue appearance and disturbance in human fetus. Early Hum Dev 1984; 10:1-11.
- Salans LB, Cushman SW, Weismann RE. Studies of human adipose tissue. Adipose cell size and number in nonobese and obese patients. J Clin Invest 1973; 52: 929-941.
- Hirsch J, Batchelor B. Adipose tissue cellularity in human obesity. J Clin Endocrinol Metab 1976; 5:299- 311.
- Ge´rard Ailhaud. Hans Hauner. Development of White Adipose Tissue. Handbook of obesity. Etiology and pathophysiology. 2006. New York.
- Flier JS. Leptin expression and action: new experimental paradigms. Proc Natl Acad Sci USA 1997; 94: 4242-4245.
- Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest 2000; 106:473-481.
- Kern PA, High adipose tissue lipoprotein lipase activity plays a causal role in the etiology of obesity. In: Angel A, Anderson H, Bouchard C, Lau D, Leiter L, Mendelson R, eds. Progress in Obesity Research: Proceedings of the Seventh International Congress on Obesity (Toronto, 1994). London: John Libbey & Company, 7:89-94.
- Carey GB. Mechanisms regulating adipocyte lipolysis. In: Richter J, ed. Skeletal Muscle Metabolism in Exercise and Diabetes New York: Plenum Press, 1998.
- Bonadonna R, Bonora E. Glucose and free fatty acid metabolism in human obesity. Relationships to insulin resistance. Diabetes Rev 1997; 5:21-51.
- Fried SK, Russell CD, Grauso NL, Brolin RE. Lipoprotein lipase regulation by insulin and glucocorticoids in subcutaneous and omental adipose tissues of obese women and men. J Clin Invest 1993; 92:2191- 2198.
- Shepherd PR, Kahn BB. Glucose transporters and insulin action: implications for insulin resistance and diabetes mellitus. N Engl J Med 1999; 341:248- 257.
- Gould GW, Holman GD. The glucose transporter family: structure, function, and tissue-specific expression. Biochem J 1993; 295:329-341.
- White MF. The IRS-signaling system: a network of docking proteins that mediate insulin and cytokines action. Recent Prog Horm Res 1998; 53:119-138.
- Czech MP, Corvera S. Signaling mechanisms that regulate glucose transport. J Biol Chem 1999; 274: 1865- 1868.
- Frevert EU, Kahn BB. Differential effects of constitutively active phosphatidylinositol 3-kinase on glucose transport, glycogen synthase activity and DNA synthesis in 3T3-L1 adipocytes. Mol Cell Biol 1997; 17:190-198.
- Saladin R, De Vos P, Guerre-Millo M. Transient increase in obese gene expression after food intake or insulin administration. Nature 1995; 377:527-529.
- Wabitsh M, Jensen PB, Blum WF. Insulin and cortisol promote leptin production in cultured human fat cells. Diabetes 1996; 45:1435-1438.
- Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR. Serum immunoreactive leptin concentrations in normalweight and obese humans. N Engl J Med 1996; 334:292-295.
- Hathout EH, Sharkey J, Racine M, Ahn D, Mace JW, Saad MF. Changes in plasma leptin during the treatment of diabetic ketoacidosis. J Clin Endocrinol Metab 1999; 84:4545-4548.
- Sinha MK, Ohannesian JP, Heiman ML, Kriauciunas A, Stephens TW, Magosin S, Marco C, Caro JF. Nocturnal rise of leptin in lean, obese, and noninsulin- dependent diabetes mellitus subjects. J Clin Invest 1996; 97:1344-1347.
- Bujalska IJ, Kumar S, Stewart PM. Does central obesity reflect, Cushing.s disease of the omentum…? Lancet 1997; 349:1210-1213.
- Renato Pasquali, Valentina Vicennati, and Uberto Pagotto. Endocrine Determinants of Fat Distribution. Handbook of obesity. Etiology and pathophysiology. 2006. New York
- Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet 2005; 365:1415-28.
- Wang CC, Goalstone ML, Draznin B. Molecular mechanisms of insulin resistance that impact cardiovascular biology. Diabetes 2004; 53: 2735-40.
- Granberry MC, Fonseca VA. Insulin resistance syndrome: options for treatment. South Med J 1999; 92:2-15.
- Greenfield JR, Campbell LV. Insulin resistance and obesity. Clin Dermatol 2004; 22:289-95.
- Ferrannini E, Natali A, Bell P, Cavallo-Perin P, Lalic N, Mingrone G. Insulin resistance and hypersecretion in obesity. European Group for the Study of Insulin Resistance (EGIR). J Clin Invest 1997; 100:1166-73.
- Cummings DE, Schwartz MW. Genetics and pathophysiology of human obesity. Annu Rev Med 2003; 54:453-71.
- DeFronzo RA. Pathogenesis of type 2 diabetes mellitus. Med Clin North Am 2004; 88:787-835 [ix].
- Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 2004; 89:2548-56.
- Ruan H, Lodish HF. Insulin resistance in adipose tissue: direct and indirect effects of tumor necrosis factor-alpha. Cytokine Growth Factor Rev 2003; 14:447-55.
- Randle PJ, Garland PB, Newsholme EA, Hales CN. The glucose fatty acid cycle in obesity and maturity onset diabetes mellitus. Ann N YAcad Sci 1965; 131:324-33.
- Lewis GF, Carpentier A, Adeli K, Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev 2002; 23:201-29.
- Ruan H, Miles PD, Ladd CM, et al. Profiling gene transcription in vivo reveals adipose tissue as an immediate target of tumor necrosis factoralpha: implications for insulin resistance. Diabetes 2002; 51: 3176-88.
- Aubert H, Frere C, Aillaud MF, Morange PE, Juhan-Vague I, Alessi MC. Weak and nonindependent association between plasma TAFI antigen levels and the insulin resistance syndrome. J Thromb Haemost 2003; 1:791-7.
- Boden G, Chen X, Ruiz J, White JV, Rossetti L. Mechanisms of fatty acidinduced inhibition of glucose uptake. J Clin Invest 1994; 93: 2438-46.
- Jenkins AB, Campbell LV. The genetics and pathophysiology of diabetes mellitus type II. J Inherit Metab Dis 2004; 27:331-47.
- DeFronzo RA, Gunnarsson R, Bjorkman O, Olsson M, Wahren J. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulindependent (type II) diabetes mellitus. J Clin Invest 1985; 76:149-55
- Saxena U, Witte LD, Goldberg IJ. Release of endothelial cell lipoprotein lipase by plasma lipoproteins and free fatty acids. J Biol Chem 1989; 264: 4349-55.
- Chandran M, Phillips SA, Ciaraldi T, Henry RR. Adiponectin: more than just another fat cell hormone? Diabetes Care 2003; 26:2442-50.
- Kinlaw GB, Marsh B. Adiponectin and HIV lipodystrophy: taking HAART. Endocrinology 2004; 145:484-6.
- Meier U, Gressner AM. Endocrine regulation of energy metabolism: review of pathobiochemical and clinical chemical aspects of leptin, ghrelin, adiponectin, and resistin. Clin Chem 2004; 50:1511- 25
- Combs TP, Berg AH, Obici S, Scherer PE, Rossetti L. Endogenous glucose production is inhibited by the adipose-derived protein Acrp30. J Clin Invest 2001; 108:1875-81.
- Yang WS, Lee WJ, Funahashi T, et al.Weight reduction increases plasma levels of an adiposederived anti-inflammatory protein, adiponectin. J Clin Endocrinol Metab 2001; 86:3815-9.
- Satoh N, Ogawa Y, Usui T, et al. Antiatherogenic effect of pioglitazone in type 2 diabetic patients irrespective of the responsiveness to its antidiabetic effect. Diabetes Care 2003; 26:2493-9.
- AldhahiW, Hamdy O. Adipokines, inflammation, and the endothelium in diabetes. Curr Diab Rep 2003; 3:293-8.
- Martin Laclaustra et al. Metabolic syndrome pathophysiology: The role of adipose tissue. Nutrition, Metabolism & Cardiovascular Diseases (2007) 17, 125-139
- Barbara Mlinar, Janja Marc, Andrej Janez, Marija Pfeifer. Molecular mechanisms of insulin resistance and associated diseases. Clinica Chimica Acta 375 (2007) 20-35
- B. Bauduceau. et al. Should we have more definitions of metabolic syndrome or simply take waist measurement? Diabetes & Metabolism. Septiembre. 2007

