
PROTEÍNA CFTR
La proteína CFTR es una cadena única polipeptídica que contiene 1 480 aminoácidos y que parece actuar como un canal de Cl- regulado por el monofosfato de adenosina (AMP) cíclico, y como su propio nombre lo indica, también regula otros canales iónicos. La forma totalmente elaborada del CFTR se encuentra en la membrana plasmática del epitelio normal. Los estudios bioquímicos indican que la mutación ∆F508 da lugar a un procesamiento inadecuado y a la degradación intracelular de la proteína CFTR. Por tanto, la ausencia de la proteína CFTR en los lugares celulares adecuados a menudo contribuye a la fisiopatología de la Fibrosis Quística. No obstante, otras mutaciones en el gen de la fibrosis quística producen proteínas CFTR completamente procesadas pero que no actúan, o lo hacen de forma parcial, en los lugares celulares adecuados.
DISFUNCIÓN EPITELIAL
Los epitelios afectados por la Fibrosis Quística muestran funciones diferentes en su estado nativo; es decir, algunos absorben volumen (vías respiratorias y epitelio intestinal), otros absorben sal pero no volumen (conductos sudoríparos), y otros secretan volumen (intestino proximal y páncreas). Dado este conjunto diverso de actividades nativas, no debe sorprender que esta patología produzca efectos muy diferentes sobre los patrones de transporte de los electrólitos y el agua. No obstante, el concepto unificado es que todos los tejidos afectados expresan una función anómala del transporte iónico. En la consulta nutricional se le pregunto a la acudiente que para fortuna del grupo investigador fue su madre biológica ¿Cómo se le había diagnosticado al paciente la fibrosis quística? Para lo cual se contó con exámenes genéticos y la prueba de Iontoforesis (electrolitos en sudor) en donde se estableció el grado de severidad de su patología y el tipo de la misma que es ∆F508.
FISIOPATOLOGÍA ESPECÍFICA DE ORGANO
PULMÓN
La clave biofísica diagnóstica de la CF es la elevación de la diferencia de potencial eléctrica transepitelial detectada en el epitelio de las vías respiratorias. La diferencia de potencial transepitelial refleja componentes tanto de la tasa de transporte iónico activo como de la resistencia al flujo de iones del epitelio superficial. El epitelio de las vías respiratorias en la Fibrosis Quística muestra tanto tasas elevadas de transporte de Na+ como disminución de la secreción de Cl-. El defecto en la permeabilidad de Cl- refleja, al menos en parte, la ausencia de la quinasa dependiente de AMP cíclico y de transporte de Cl- regulado por la proteincinasa C, que está mediado por las funciones de canal de Cl- de CFTR. Una observación importante es que existe un canal Ca2+ de Cl- «alternativo» expresado en el epitelio de las vías respiratorias. Este canal de Cl- es diferente de CFTR y está regulado por las concentraciones de Ca2+ intracelular. Este canal puede sustituir al CFTR en lo que se refiere al transporte neto de Cl- y puede constituir un objetivo terapéutico potencial.
La absorción elevada de Na+ es una característica habitual del epitelio de las vías respiratorias en la Fibrosis Quística. Las anomalías del transporte de Na+ no son una característica general del fenotipo epitelial de la Fibrosis Quística y parecen limitadas a los epitelios que absorben volumen. Estudios recientes han demostrado que la absorción incrementada de Na+ refleja una segunda función del CFTR: actúa como inhibidor tónico del canal epitelial de este ion. Aún no ha podido identificarse el mecanismo molecular que media esta acción del regulador transmembrana de la fibrosis quística.
La depuración del moco parece ser un mecanismo de defensa primario innato de las vías respiratorias contra la infección por las bacterias inhaladas. Las vías respiratorias normales varían las tasas de absorción de Na+ y de secreción de Cl- activas a fin de ajustar el volumen de líquido (agua) sobre las superficies de las vías respiratorias para la eliminación eficiente del moco. La hipótesis central de la fisiopatología de las vías respiratorias en caso de Fibrosis Quística considera que la tasa anormalmente elevada de absorción de Na+ y la tasa muy baja de excreción de Cl- reducen el contenido de sal y agua del moco y agotan el volumen del líquido periciliar. Tanto el espesamiento del moco como el agotamiento del líquido periciliar producen adherencia del primero a la superficie de las vías respiratorias. La adherencia del moco tiene como consecuencia incapacidad para eliminarlo normalmente de las vías respiratorias por mecanismos ciliares o dependientes del flujo de aire (tos). Se inclinan a favor de esta hipótesis datos recientes obtenidos de modelos de cultivos de Fibrosis Quística de células humanas.
La infección que caracteriza a las vías respiratorias de las Fibrosis Quística abarca a la capa de moco en vez de invadir la capa epitelial o las propias vías. La predisposición única de éstas en caso de Fibrosis Quística a la infección crónica por Staphylococcus aureus y Pseudomonas aeruginosa es compatible con la incapacidad para eliminar el moco, pero se ha sugerido que pueden contribuir también anormalidades aún no definidas del líquido de la superficie de estas vías a la selección de estos microorganismos. Hace poco se demostró que la tensión de O2 reducida en el moco de la Fibrosis Quística antes de establecerse la infección, y particularmente después de haberlo hecho, puede ser el factor que selecciona a estas bacterias y ser el determinante de su fenotipo. Por este motivo, tanto la estasis como la hipoxia del moco pueden contribuir a la proclividad de Pseudomonas a crecer en colonias de biopelícula dentro de las placas de moco adheridas a las superficies de las vías respiratorias afectadas por la fibrosis quística.
TUBO DIGESTIVO
Los efectos digestivos de la Fibrosis Quística son diversos. En el páncreas exocrino parece que la ausencia del canal CFTR del Cl- en la membrana apical del epitelio ductal pancreático altera la función de un intercambiador de Cl–HCO3- de dicha membrana para secretar HCO3 y Na+ (por un proceso pasivo) al conducto. La incapacidad para secretar Na+-HCO3- y agua da lugar a la retención de enzimas en el páncreas y finalmente a la destrucción de casi todo el tejido pancreático. En la Fibrosis Quística, el epitelio intestinal, debido a la falta de secreción de Cl- y agua, muestra una alteración de la capacidad para limpiar las mucinas secretadas y otras macromoléculas de las criptas intestinales. La secreción reducida de líquidos mediada por el CFTR puede agudizarse por una absorción excesiva de los mismos en el intestino distal, que refleja las anomalías en la regulación de la absorción de Na+ mediada por el CFTR (ambas alteraciones se deben a los canales de Na+ y posiblemente a otros transportadores de Na+, como los intercambiadores Na+-H+). Las dos disfunciones conducen a la desecación del contenido intraluminal y la obstrucción del intestino delgado y grueso. En el sistema hepatobiliar, la defectuosa secreción de Cl- y agua en los conductos hepáticos ocasiona una retención de las secreciones biliares y cirrosis biliar local, así como una proliferación de conductos biliares en aproximadamente el 25 a 30% de los pacientes con Fibrosis Quística. La incapacidad del epitelio de la vesícula biliar en la Fibrosis Quística para secretar sal y agua puede originar tanto colecistitis crónica como colelitiasis.
GLÁNDULAS SUDORÍPARAS
Los pacientes con Fibrosis Quística secretan volúmenes casi normales de sudor al ácino glandular, pero son incapaces de absorber NaCl del sudor durante el paso de éste por el conducto glandular, porque las células epiteliales ductales no pueden absorber Cl-. Por este motivo la prueba de la Iontoforesis es el mejor testigo de la patología y el compromiso de la misma.
MANEJO NUTRICIONAL PARA LA FIBROSIS QUÍSTICA
La interacción de numerosos factores para regular el balance energético es claramente lo más complicado. La mayoría de pacientes con fibrosis quística pueden mantener el peso corporal por periodos cortos aunque el reporte de la energía consumida este por debajo de los valores óptimos, por ejemplo la mayoría de pacientes requieren de 120 a 150% del porcentaje de adecuación en la ingesta de sus recomendaciones diarias. Sin embargo es necesario mantener a estos pacientes en estricta vigilancia nutricional. El cuidado debe ser individualizado, ya que estos pacientes varían mucho en cuanto a sus necesidades nutricionales. Los reportes del consenso norteamericano muestran en la siguiente tabla las distintas categorías de pacientes con fibrosis quística en cuanto al tipo de tratamiento nutricional que deben recibir:
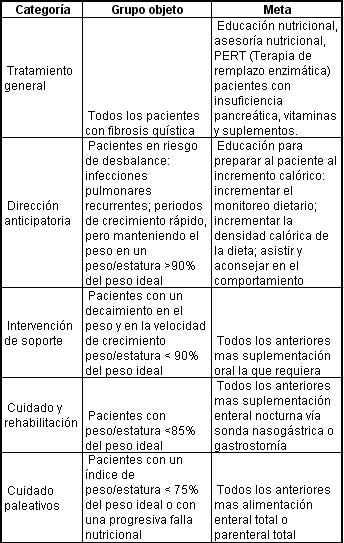
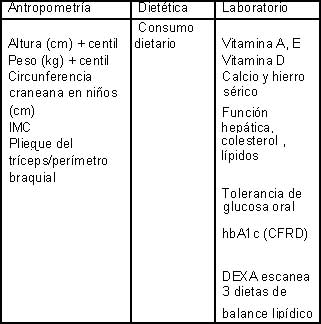
Los objetivos nutricionales para el cuidado y el tratamiento son: I. mantener o mejorar el estatus nutricional en los adultos (y el crecimiento en los niños), y II. Prevenir del desbalance nutricional como las deficiencias en vitaminas y minerales. Es por lo tanto uno de los factores más importantes en los pacientes con fibrosis quística el revisar regularmente el estatus nutricional asesorar sobre este y tener en cuenta esta información. Los intervalos para hacer las mediciones antropométricas básicas no debe exceder los tres meses, y el control nutricional completo consistente en todas las medidas antropométricas, pruebas de apoyo diagnóstico y quizá pruebas de imagen no deben sobrepasar un año. La imposibilidad para mantener el peso, y la velocidad de crecimiento, requiere de la oportuna respuesta medica que puede variar en el aseguramiento del mismo por el no reconocimiento o el no adecuado tratamiento de las infecciones respiratorias, se requiere el empleo de un especifico acercamiento nutricional donde se incluya (PERT) sonda nasogástrica o gastrostomía, suplementos y hasta en algunos casos complicados y un poco raros la nutrición parenteral total. La deficiencia de cualquier micronutriente debe ser corregida de inmediato. Un avance en de las recomendaciones actuales, es el de manejar la edad ajustada por el percentil de índice de masa corporal, ya que este es un indicador más sensible de la condición nutricional que los percentiles del indicador peso/estatura. Los pacientes con índice de masa corporal por debajo el percentil 25 se encuentran en riesgo y deben ser tenidos en cuenta para iniciar una intervención. Mientras que los que están por debajo del percentil 10 son clasificados en falla nutricional y deben ser tratados de inmediato. En muchos centros clínicos los pacientes con fibrosis quística son vistos por los dietistas en cada visita; esto es particularmente importante en el caso de que se pueden diagnosticar nuevos casos de fibrosis quística especialmente en niños y en adolescentes, en donde el mantener la velocidad de crecimiento es la prioridad.
Asistencia nutricional de rutina en los pacientes con fibrosis quística
En el caso de diagnosticar esta patología tempranamente cuando los menores todavía están en buenas condiciones, como por ejemplo en el «screening» neonatal, se recomienda esencialmente que se continúe con la lactancia materna. La leche humana tienen el óptimo balance de aminoácidos y ácidos grasos esenciales, también contiene lipasa y amilasa que compensan las secreciones pancreáticas disminuidas. Mejor aún, la presencia de inmunoglobulinas, lactoferrina, factor de crecimiento epidérmico y lisozimas hacen de esta leche una protección natural contra las infecciones tan acuñadas a esta patología. El contenido de taurina que es necesaria para la síntesis de ácidos biliares se ha demostrado que mantiene y mejora la absorción de las grasas. Los infantes no alimentados con lactancia materna, pueden cursar con fórmulas lácteas con una adecuada terapia enzimática, las que contiene proteína hidrolizada no tienen ventaja sobre las demás fórmulas como el común denominador lo manifiesta.
Los adultos que tiene fibrosis quística, también deben estar bajo un frecuente cuidado nutricional, idealmente cada seis meses. La pérdida de peso requiere de un apropiado cuidado y debe ser tratado de inmediato. Los periodos nutricionales de mayor crisis donde se requiere una vigilancia constante son: adolescencia donde se exige una terapia médica y nutricional adecuada para la edad. En la mayoría de los pacientes es frecuente encontrar dietas hiperproteicas e hipocalóricas ya que así se logra mantener bien nutrido al paciente por algunos años. Un peligro de esto es el tipo de dieta deficiente en fibra, que se asocia con dolor abdominal y síntomas colonicos, esto no es general en todos los pacientes y varía mucho con las costumbres regionales y patrones alimentarios.
Mantener un normal estatus nutricional es la labor más importante en el trato de un paciente con fibrosis quística y se requiere de un equipo multidisciplinario para el tratamiento, este se va haciendo más difícil con el aumento en la edad, con la colonización crónica con Pseudomonas aeruginosa y con las progresivas crisis respiratorias y su posterior falla. El primer paso del programa inicia con la carga nutricional y suplementaria por vía oral; usualmente se requiere nutrición enteral y en muy raras ocasiones nutrición parenteral. Las recomendaciones calóricas y de suplementación deben ser diseñadas según el individuo y las manifestaciones de la patología.
Puntos prácticos
- Vigilancia nutricional regular necesaria = anticipar el problema
- Muchos pacientes requieren PERT, pero antes se debe justificar la presencia de la deficiencia pancreática
- La carga calórica debe aumentarse, pero se debe tener en cuenta el apetito del paciente, ya que este puede ser muy pobre
- Los suplementos nutricionales pueden ser efectivos cuando se inician de manera temprana cuando hay declinamiento nutricional
- Los suplementos comunes son más efectivos que los más caros
- Las deficiencias de vitaminas y minerales son evidentes, pero en algunas formas no son fácilmente descubiertos en la clínica y se hace necesario realizar pruebas bioquímicas
- La fibrosis quística asociada con diabetes usualmente requiere el uso de insulina
BIBLIOGRAFÍA
- BORON, Walter. BOULPAEP, Emile. Medical Physiology. SAUNDERS. ElServier Science. USA. 2003.
- MAHAN, K. ESCOTT, Stump. Krause’s Food, nutrition and diet therapy. 11th edition. Saunders. Evolve. 2004.
- SHILS, Maurice. OLSON, James. SHIKE, Moshe. ROSS, Catherine. Modern nutrition in health and disease. LIPPINCOTT WILLIAMS & WILKINS. USA. 1999.
- STIPANUK, H, Martha. Biochemical and Physiological Aspects of Human Nutrition. Saunders Company. 2000.
- DODGE, John. TURCK, Dominique. Cystic fibrosis: Nutritional consequences and management. Best practice and research Clinical Gastroenterology. Vol 20 nº 3 pp 531-546, 2006.
- Ramsey BW, Farrell PM, Pencharz P et al. Nutritional assessment and management in cystic fibrosis: a consensus report. Am J Clin Nutr 1992; 55: 108-116.
- Sinaasappel M, Stern M, Littlewood J et al. Nutrition in patients with cystic fibrosis: a European. Consensus. J Cyst Fibros 2002; 1: 51-75.
