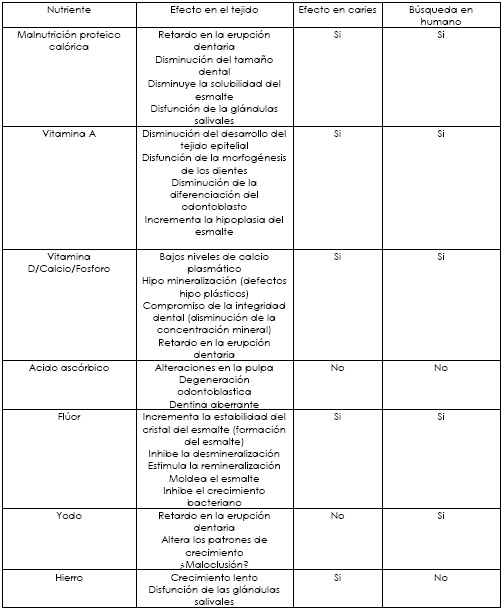
Fenilcetonuria
Fisiología de la conversión de Fenilalanina a Tirosina
La mayor tasa catabólica de fenilalanina a tirosina ocurre en el hígado, pero la tirosina es muy importante como el precursor para la síntesis de muchos compuestos en otros tejidos.
La fenilalanina es convertida a tirosina por la enzima fenilalanina hidroxilasa, que es un tetrahidrobioterin dependiente que mezcla la función de oxidar e hidroxilar la fenilalanina en el carbono 4 del anillo aromático.Esta reacción es el primer paso en la degradación de la fenilalanina y también es el paso que permite que este aminoácido sea el precursor dietario de la tirosina.La reacción reversa no ocurre y la tirosina no puede ser reemplazada totalmente por la fenilalanina de la dieta, solo aproximadamente un 50% del total de la fenilalanina + tirosina puede ser proveída como tirosina.
La conversión de fenilalanina hasta tirosina, desde el catabolismo de la fenilalanina está regulada por cambios en la actividad de la fenilalanina hiroxilasa, que es regulada a su vez por un estado de fosforilación. La fosforilación del residuo de seril en el dominio regulador de la enzima activa la enzima fenilalanina hidroxilasa por un incremento en su actividad específica. Esta fosforilación hace que la enzima se exprese en gran medida.
Lo que responde al incremento de la fenilalanina consumida en la dieta (alta en proteína) o por un incremento en los niveles de glucagón circulante (hormona catabólica) que actúan liberando cantidades enormes de dicha enzima. La tasa de la fosforilación de la enzima es catalizada por AMP cíclico dependiente de protein kinasa y calcio / calmodulina dependiente de protein kinasa.
Los anteriores procesos servirán de base para entender la fisiopatología del error innato del metabolismo de este aminoácido.
Base Fisiopatológica
Fenilcetonuria: Básicamente se trata de un error innato del metabolismo como se describe en la actualidad, un defecto en el gen que codifica para la enzima fenilalanina hidroxilasa que imposibilita la conversión del aminoácido fenilalanina a tirosina. Esta enfermedad tiene como rasgo principal la herencia genética autosómica recesiva, es decir, los padres son portadores de genes defectuosos y al ser traspasados de ambos progenitores la enfermedad se expresa.
La incidencia es de uno de cada 16.000 nacimientos padecen del error en los Estados Unidos. Gracias a las pruebas de diagnóstico en neonatos es fácilmente detectable mediante los cribados metabólicos habituales. Se conoce desde 1934, año en que fue descrita por el noruego Ivar Asbjorn Folling, luego en 1937 Penrose y Quastel sugieren que sea denominada la enfermedad como Fenilcetonuria. En 1947 Jervis observó que cuando se daba fenilalanina a individuos sanos en estos se producía un aumento de otra sustancia llamada tirosina.
Pero cuando suministraba fenilalanina a individuos fenilcetonúricos, no presentaban tal elevación. Los avances en el tratamiento no se iniciaron hasta 1953, año en que fueron publicados trabajos en los que se demostraba la efectividad de la dieta baja en fenilalanina.
La causa de la enfermedad es la carencia de la enzima fenilalanina hidroxilasa (FAOH) o de la dihidropterina reductasa (DPHR) (también llamada tirosina hidroxilasa). Ambas enzimas son responsables de la hidroxilación del aminoácido fenilalanina en la reacción que produce tirosina. Por ello, el defecto o falta de alguna de ellas determina un incremento de la concentración sanguínea de fenilalanina al impedirse su transformación en tirosina. También se aumenta la transaminación de la fenilalanina como vía metabólica alternativa, y asimismo se acumulan los metabolitos fenilpiruvato, fenilactato y fenilacetato.
Los niveles de tirosina comienzan a ser peligrosamente inadecuados, y la fenilalanina se convierte en fenilpiruvato que es un compuesto dañino en el desarrollo del cerebro. El fenilpiruvato es un neurotóxico que afecta gravemente al cerebro durante el crecimiento y el desarrollo. Los efectos de la acumulación de este neurotóxico causan oligofrenia fenilpirúvica, caracterizada por un cociente intelectual inferior a 20. Los primeros meses de vida, los niños que padecen esta enfermedad parecen estar sanos. Entre los tres y los seis meses pierden el interés por el entorno, y al año se evidencia un retraso importante en su desarrollo.
Los síntomas suelen ser retraso psicomotor, cuadros psicóticos de tipo autista, convulsiones, síndrome de West, convulsiones generalizadas y un eczema facial muy rebelde. Por lo general su desarrollo físico es bueno, tienden a tener el cabello más claro que sus hermanos, piel clara, y presentan un olor característico a paja mojada.
Debido a que la fenilalanina está comprometida de forma indirecta en la producción de la melanina, el pigmento responsable del color de la piel y del cabello, los niños con fenilcetonuria usualmente presentan los rasgos anteriormente descritos como un cutis más claro que el de los hermanos no afectados. Estos niños presentan también el olor similar al del ratón que resulta de la acumulación de ácido fenilacético y se puede sentir en el aliento, en la piel y en la orina si la condición no ha sido tratada de inmediato desde el nacimiento o cuando se consumen alimentos que contienen fenilalanina.
La fenilcetonuria es una enfermedad que se puede tratar y detectar fácilmente a través de un examen de sangre simple. La mayoría de los estados (en Estados Unidos) exigen un examen de tamizaje para todos los recién nacidos, que generalmente se hace con una punción en el talón poco tiempo después del nacimiento.
El papel del nutricionista en el tratamiento nutricional involucra la restricción del sustrato fenilalanina y la consiguiente suplementación del producto tirosina. La gran mayoría de afectados son infantes que exhiben esta deficiencia, los restantes cerca del 3% tienen defectos asociados a las vías metabólicas implicadas. La terapia con bajas dosis de fenilalanina no previene el deterioro neurológico presente en los desórdenes asociados a las vías. La fenilalanina se encuentra en cantidades significativas en alimentos como la leche, los huevos y otros alimentos comunes.
Además, se encuentra en el edulcorante Nutrasweet (aspartame), razón por la cual los productos que contengan aspartame se deben evitar en las dietas de los niños con esta enfermedad. Lofenalac® es una leche en polvo infantil especial para niños con fenilcetonuria que se puede usar durante toda la vida como fuente de proteína con un contenido extremadamente bajo en fenilalanina y balanceado para los aminoácidos esenciales restantes.
Las mujeres adultas portadoras de fenilcetonuria que tienen planes de quedar embarazadas también deben seguir una dieta estricta baja en fenilalanina antes de quedar embarazadas y durante todo el embarazo. Se recomienda la asesoría genética para los futuros padres con antecedentes familiares de fenilcetonuria. El estado portador para esta enfermedad se puede detectar por medio de pruebas enzimáticas y, además, se puede hacer un diagnóstico prenatal de dicha afección.
DIAGNOSTICO Y CRITERIOS DE APARICIÓN
Todos los estados de búsqueda en los neonatos son programados por buscar cualquier desorden metabólico. Los ensayos de la inhibición bacteriana de Guthrie, y los realizados en la sangre, son los más usados como test de búsqueda. La Academia Americana de Pediatrita recomienda que los neonatos que salgan positivos en dichas pruebas deben ser sometidos a tanto a pruebas cualitativas como cuantitativas.
El diagnóstico para dictaminar fenilcetonuria incluye valores superiores o que excedan de 6 a 10mg/dl en sangre de fenilalanina y valores inferiores o por debajo de 3mg/dl de tirosina. Como se comentó anteriormente el desarrollo cognoscitivo del menor se ve comprometido si no se realiza un adecuado cuidado nutricional, la terapia nutricional es muy importante ya que se logra un control bioquímico funcional de la disfunción enzimática.
Los infantes con fenilcetonuria no manifiestan ningún signo clínico de la anormalidad en el periodo postnatal, la edad del infante es el factor relevante en la terapia nutricional y esto depende de la efectividad del proceso de búsqueda del desorden metabólico.
Las ventajas de una detección a tiempo de esta irregularidad ayudan al profesional nutricionista en realizar la terapia a tiempo y prevenir el daño al desarrollo del sistema nervioso. Los individuos que no reciben una terapia nutricional a tiempo presentan muchos retardos en las funciones mentales (con un IQ entre 40) en contraste de quienes reciben la terapia a tiempo que mantiene dichas funciones normales, en un rango de intelecto como cualquier otro.
TERAPIA MÉDICA NUTRICIONAL EN INFANTES
FORMULA
Una dieta libre de fenilalanina dentro del plan dietario es necesaria para seleccionar la formula donde estas son formulas con fuentes proteicas donde es removido dicho aminoácido. En general la fuente de proteína de la formula son aminoácidos con isómeros L donde se omite el aminoácido esencial para el cual la enzima no actúa. La fuente de carbohidratos es proveniente de jarabes de maíz, almidón modificado de tapioca, sucrosa y un hidrolizado de almidón de maíz.
La grasa se obtiene de una variedad de aceites. Estas fórmulas libres de fenilalanina son suplementadas con fórmulas regulares lácteas o con leche materna durante la infancia y con leche de vaca en la infancia temprana para proveer una proteína de alto valor biológico. Para mantener el control del soporte metabólico de una manera efectiva, tanto las fórmulas como la comida que va a hacer consumida debe ser en porciones equivalentes en todo el transcurso del día.
CONTROL DE FENILALANINA EN SANGRE
Las concentraciones de fenilalanina deben ser revisadas regularmente, dependiendo de la edad y el estatus de salud del menor para estar seguros de que el rango se mantiene de 2 a 6 mg/dl. Los alimentos que contienen fenilalanina pueden ser tolerados si estos se calculan en el tratamiento nutricional del paciente y no alteraran las concentraciones plasmáticas normales y mantendrán el control bioquímico.
La tasa de crecimiento y desarrollo del menor deben ser monitoreadas en especial el desarrollo mental. Todo este trabajo debe ser una coordinación de un equipo interdisciplinario donde se encuentra el nutricionista como autor intelectual del proceso del ambiente favorable donde se desarrollara el menor.
Las elevaciones de fenilalanina en sangre generalmente son causadas por un consumo elevado de este aminoácido o por catabolismo de tejidos. El consumo de fenilalanina en exceso más de la cantidad requerida para el crecimiento se acumula en la sangre.
Un consumo deficiente de energía, un trauma, enfermedad o infección llevan a una baja en las proteínas plasmáticas con la consecuente liberación de aminoácidos a la sangre incluyendo este aminoácido. Prevenir el catabolismo en dichas condiciones, seleccionar una fórmula adecuada, seleccionar una dieta para pacientes en estos estados hipermetabólicos es una tarea complicada pero que le concierne al equipo de trabajo y al nutricionista de gran manera.
La mejor recomendación para toda la vida ya que no solo el desarrollo intelectual se ve comprometido en niños en desarrollo, sino también una serie de alteraciones comportamentales en adultos y adolescentes, perdidas de atención, disminución de las capacidades de aprehensión se manifiestan con claridad sino se controlan los niveles de fenilalanina durante toda la vida, la terapia nutricional debe continuar en cada momento si se quiere mantener las funciones cognitivas normales.
CUIDADO NUTRICIONAL EN GESTANTES FENILCETONURICAS
Una mujer embarazada con concentraciones elevadas de fenilalanina coloca en riesgo al feto porque amplifica el transporte de aminoácidos a través de la placenta.
El feto está expuesto a casi dos veces más a este aminoácido en comparación con las gestantes normales. Los bebes de madres con elevadas concentraciones de fenilalanina en sangre tiene un incremento en el padecimiento de defectos cardiacos, retraso del crecimiento, microcefalia y retraso mental.
El manejo nutricional y la terapia de este tipo de mujeres con hiperfenilalaninemia son muy complejos. El cambio de la fisiología de las mujeres embarazadas y el cambio en las demandas nutricionales dificultan el monitoreo y la correcta precisión de mantener apropiadamente las bajas concentraciones de fenilalanina. Aun con una atención meticulosa del consumo de este aminoácido y de las concentraciones del mismo en sangre los requerimientos del mismo en el desarrollo del infante dificultan el asegurar un bienestar tanto para la madre como para él bebe.
El riesgo de un desarrollo anormal del feto aun con el cuidado terapéutico dietario manteniendo las concentraciones plasmáticas de este aminoácido de 1 a 5 mg/dl es importante para considerar un embarazo para una mujer con fenilcetonuria.
BIBLIOGRAFÍA
TEXTOS
- MAHAN, K. ESCOTT, Stump. Krause’s
- STIPANUK, H, Martha. Biochemical and Physiological Aspects of Human Nutrition. Saunders Company. 2000.
ARTICULO BASE
- Okano Y, Hase Y, Kawajiri M, Nishi Y, Inui K, Sakai N, Tanaka Y, Takatori K, Kajiwara M, Yamano T. In vivo studies of phenylalanine hydroxylase by phenylalanine breath test: diagnosis of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Department of Pediatrics, Osaka City University Graduate School of Medicine, 1-4-3 Asahimachi, Abeno-ku, Osaka 545-8585, Japan. okano@med.osaka-cu.ac.jp. 2004 Nov; 56(5):714-9. Epub 2004 Aug 19.